
Amyotrophic lateral sclerosis (ALS) is a rare neurological disease affecting more than 500.000 people worldwide, with an average life expectancy of about 2-5 years from the time of diagnosis1. ALS is characterized by muscle atrophy and respiratory failure. A key pathological hallmark of ALS is the accumulation of proteins, such as TAR DNA-binding protein 43, or fused in sarcoma (FUS) that leads to the loss of motor neurons. Most ALS cases are sporadic (~90%), and genetic studies have linked more than 30 genes to familial cases2.
Until recently, there were only three FDA approved drugs for ALS on the market. The first medication approved for ALS patients was Rilutek, in 1995, which prolongs survival but does not reverse nerve damage. This was followed by Nuedexta in 2011, which targets specific ALS side effects. In 2017, after more than 13 years of development, Edaravone was approved. It reduces oxidative stress and may attenuate motor neuron degeneration, but it seems to be only effective on a well-defined group of patients with early ALS3. In 2022, the FDA approved Relyvrio, a combination of two drugs, shown to reduce the rate of decline of daily functioning and associated with longer overall survival4.
Given the lack of understanding of the onset of ALS for most patients, patient-specific iPSC-derived motor neurons offer the opportunity to bring patients' genome and phenotypes into a dish to identify lead therapeutic candidates with agnostic screenings. As an example, Retigabine, an FDA-approved potassium channel inhibitor, effectively blocked the pathological hyperexcitability and improved neuron survival in iPSC-derived motor neurons from ALS patients5. Based on the relevance of this phenotype in ALS pathology and the absence of relevant animal models, Retigabine was directly moved into a phase II clinical trial6. In another drug-repositioning study, three lead candidates were identified for their ability to rescue neurite retractions, hyperexcitability and autophagy levels in iPSC-derived motor neurons from ALS patients7. The three candidates are currently being investigated in clinical trials 6. One of these 3 compounds, ropinirole, was screened on 32 patient-derived motor neurons, being able to discriminate specific patient responses to the drug 72 and it is now being manufactured to be commercialize in Japan.
Phenotypic screens with iPSC-derived neurons from ALS patients also offer the possibility to validate known and discover novel targets with high confidence. A study with a multistep screening of 2899 compounds identified 13 targets that modulate motor neuron excitability and found a new class of candidate drugs, agonists to the dopamine D2 receptor, with the ability to reduce hyperexcitability9.
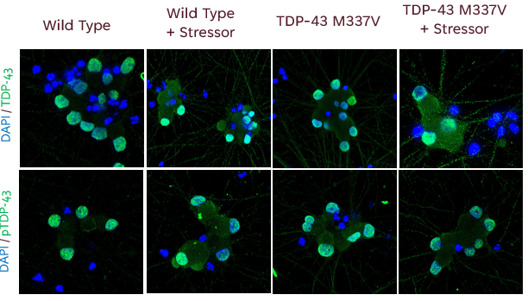 Utilizing in vitro cell models that mimic specific ALS mutations and phenotypes can help identify unknown effects of mutations and potential targets for drug development. In the long run, identifying these targets will help to predict the efficacy and effect of new therapeutic candidates with higher confidence.
Utilizing in vitro cell models that mimic specific ALS mutations and phenotypes can help identify unknown effects of mutations and potential targets for drug development. In the long run, identifying these targets will help to predict the efficacy and effect of new therapeutic candidates with higher confidence.
Ncardia recently developed an iPSC-derived ALS disease model, using motor neurons with a TDP-43 point mutation (iCell® Motor Neurons, 01279 from FUJIFILM Cellular Dynamics, Inc.) to quantify disease relevant phenotypes: Mis-localization of TDP-43, Aggregation of TDP-43, Mis-splicing of STMN2, Electrophysiological dysfunction. Explore the applications in our case study.
References
[1] Han C, Chaineau M, Chen CXQ, Beitel LK, Durcan TM. Open science meets stem cells: A new drug discovery approach for neurodegenerative disorders.
Front Neurosci. 2018;12(FEB):47. doi:10.3389/FNINS.2018.00047/BIBTEX.
[2] Pasteuning-Vuhman S, de Jongh R, Timmers A, Pasterkamp RJ. Towards Advanced iPSC-based Drug Development for Neurodegenerative Disease.
Trends Mol Med. 2021;27(3):263-279. doi:10.1016/J.MOLMED.2020.09.013
[3] Hardiman O, van den Berg LH. Edaravone: a new treatment for ALS on the horizon? Lancet Neurol. 2017;16(7):490-491. doi:10.1016/S1474-
4422(17)30163-1
[4] FDA news release (2022). FDA Approves New Treatment Option for Patients with ALS. Accessed December 16, 2022. https://www.fda.gov/news-events/press-announcements/fda-approves-new-treatment-option-patients-als
[5] Wainger BJ, Kiskinis E, Mellin C, et al. Intrinsic Membrane Hyperexcitability of Amyotrophic Lateral Sclerosis Patient-Derived Motor Neurons. Cell Rep.
2014;7(1):1-11. doi:10.1016/J.CELREP.2014.03.01
[6] Wainger BJ, Macklin EA, Vucic S, et al. Effect of Ezogabine on Cortical and Spinal Motor Neuron Excitability in Amyotrophic Lateral Sclerosis: A Randomized Clinical Trial. JAMA Neurol. 2021;78(2):186-196. doi:10.1001/JAMANEUROL.2020.4300
[7] Okano H, Yasuda D, Fujimori K, Morimoto S, Takahashi S. Ropinirole, a New ALS Drug Candidate Developed Using iPSCs. Trends Pharmacol Sci.2020;41(2):99-109. doi:10.1016/J.TIPS.2019.12.002
[8] Fujimori K, Ishikawa M, Otomo A, et al. Modeling sporadic ALS in iPSCderived motor neurons identifies a potential therapeutic agent. Nat Med 2018 2410. 2018;24(10):1579-1589. doi:10.1038/s41591-018-0140-5
[9] Huang X, Roet KCD, Zhang L, et al. Human amyotrophic lateral sclerosis excitability phenotype screen: Target discovery and validation. Cell Rep. 2021;35(10):109224. doi:10.1016/J.CELREP.2021.109224/ATTACHMENT/C8F909D5-7D7E-44A3-979E-3543B46F44AE/MMC4.XLSX